

Dr Paolo Gallipoli
MB ChB, PhD, MRCP, FRCPath
Clinical Reader in Experimental Haematology
Group Leader
TwitterResearch Focus
My research interests focus on mechanisms of disease initiation and maintenance and the identification and validation of novel therapeutic targets in myeloid leukaemias. Specifically I study the role of adaptive responses to current therapies, including novel targeted therapies, in several subtypes of myeloid leukaemias and at leukaemic stem cell level, with a specific focus on the role of metabolic adaptations, as a mechanism of resistance not driven by genetic mutations.
Key Publications
- Immunodeficient NBSGW mouse strain allows chemotherapy modeling in AML patient-derived xenografts. Hemasphere. (2024) 8(1):e28. PMID 38434525
- Mannose metabolism inhibition sensitizes acute myeloid leukaemia cells to therapy by driving ferroptotic cell death. Nature Communications (2023) 14(1):2132. PMID: 37059720
- C/EBPα Confers Dependence to Fatty Acid Anabolic Pathways and Vulnerability to Lipid Oxidative Stress-Induced Ferroptosis in FLT3-Mutant Leukemia. Cancer Discovery. 13(7):1720-1747. PMID: 37012202
- Glutaminolysis is a metabolic dependency in FLT3ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood (2018) 131(15):1639-1653. PMID: 29463564
- JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood (2014) 124(9):1492-501. PMID: 24957147
Major Funding
- 2023 - 2026- Barts Charity project grant, £320,237
- 2023 - NIHR Academic Clinical Fellowship, "Identification of novel therapies in acute myeloid leukaemia (AML)", approx. £200,000
- 2022 - Cancer Research UK City of London Development Fund 2022, “Identification of the metabolic determinants of the anti-leukemic effects of stearoyl-coA desaturase (SCD) inhibition”, £ 25,000
- 2022 - Lady Tata Memorial Trust International Award (awarded to Dr Dembitz Postdoctoral Research Assistant in the lab), “Mechanisms of anti-leukemic effects of stearoyl-coA desaturase (SCD) inhibition”, £33,000
- 2021-2025 - CRUK City of London PhD Scholarship, "Exploring adaptive resistance to targeted therapy in AML," £146,622
- 2019-2024 - CRUK Advanced Clinician Scientist Fellowship, "Identification and characterisation of metabolic pathways as determinants of drug resistance in acute myeloid leukaemia," £1,427,558
Other Activities
- 2024 - Research Foundation Flanders (FWO) – Postdoctoral Fellowship Panel
- 2023 - Research Foundation Flanders (FWO) – PhD Fellowship Panel
- 2021 - ASH Global Research Award Panel
- Member of the ASH Global Research Award Grant Subcommittee
- Member of UK, National Cancer Research Institute (NCRI) CML and AML clinical and scientific study group
Themes/Keywords
Research
Acute myeloid leukaemia (AML) is the most prevalent acute leukaemia in adults and a cancer of unmet need with long-term survival rates of less than 30%. Sequencing and mechanistic studies have improved our understanding of the biology of several subtypes of myeloid leukemics. This has in turn resulted in the development of more targeted and scientifically validated therapies. However, the overall treatment outcomes, even with the introduction of novel agents, remain suboptimal for most patients, mainly as a result of disease relapse. AML arises in a haematopoietic stem or progenitor cell, which has the ability to self-renew, following the acquisition of recurrent driver mutations. This cell of origin, usually named the Leukaemia Stem Cell (LSC), represents the reservoir for relapse due to its inherent or acquired resistance to current therapies. Therefore, an improved understanding of the molecular mechanisms causing disease relapse, particularly at the LSC level, is required to improve patient outcome.
My research interests focus on mechanisms of disease initiation and maintenance and the identification and validation of novel therapeutic targets in myeloid leukaemia and studies the role of adaptive responses to current therapies, including novel targeted therapies, in several subtypes of myeloid leukaemias and at LSC level, with a specific focus on the role of metabolic adaptations, as a mechanism of resistance not driven by genetic mutations. Metabolic alterations are a hallmark of AML, and are often responsible for the development of chemoresistance arising from the bottleneck of extreme metabolic stress during intensive chemotherapy. Several metabolic pathways have been reported by our groups and others to be essential for leukaemic cell maintenance and resistance to therapy. For example we have shown reliance on glutamine and other energy sources from bone marrow niche to feed tricarboxylic acid cycle activity, oxidative phosphorylation and glutathione generation to drive therapy resistance and the role of mannose metabolism in modulating the ability of AML cells to switch the majority of energy dependence on fatty acid oxidation in response to standard and novel therapies.
In the laboratory, we use a combination of forward genetic screening, functional and mechanistic studies to characterise the clonal dynamics in leukaemic cell populations under therapeutic stress, characterise the mechanisms leading to therapy resistance and identify novel therapeutic vulnerabilities to be targeted in combination with standard therapies. We are also interested in the bidirectional cross-talk between altered metabolism and aberrant signalling and transcriptional programmes and its role in the establishment of myeloid leukaemias. We specifically study how metabolic intermediates interact with transcriptional programmes/altered signalling in AML and conversely how specific driver mutations impact on cellular metabolism to enable leukemic transformation.
We use cell lines, primary patient samples and murine models and study the functional effects of targeting novel vulnerabilities by combining complementary approaches such as RNAseq, Proteomics, Phosphoproteomics, Metabolomics, Lipidomics, in vivo imaging and drug screening.
Key words: signalling, transcription, patient samples, in vivo models, microenvironment
Ongoing projects:
- Role of mannose metabolism in AML therapy resistance to standard and novel therapies
- Role of mannose metabolism in leukaemia stem cell function
- Role of lipid biosynthesis in AML establishment and maintenance
- Cross-talk between microenvironment and AML cells in FLT3 and IDH mutant AML and how it leads to therapy resistance
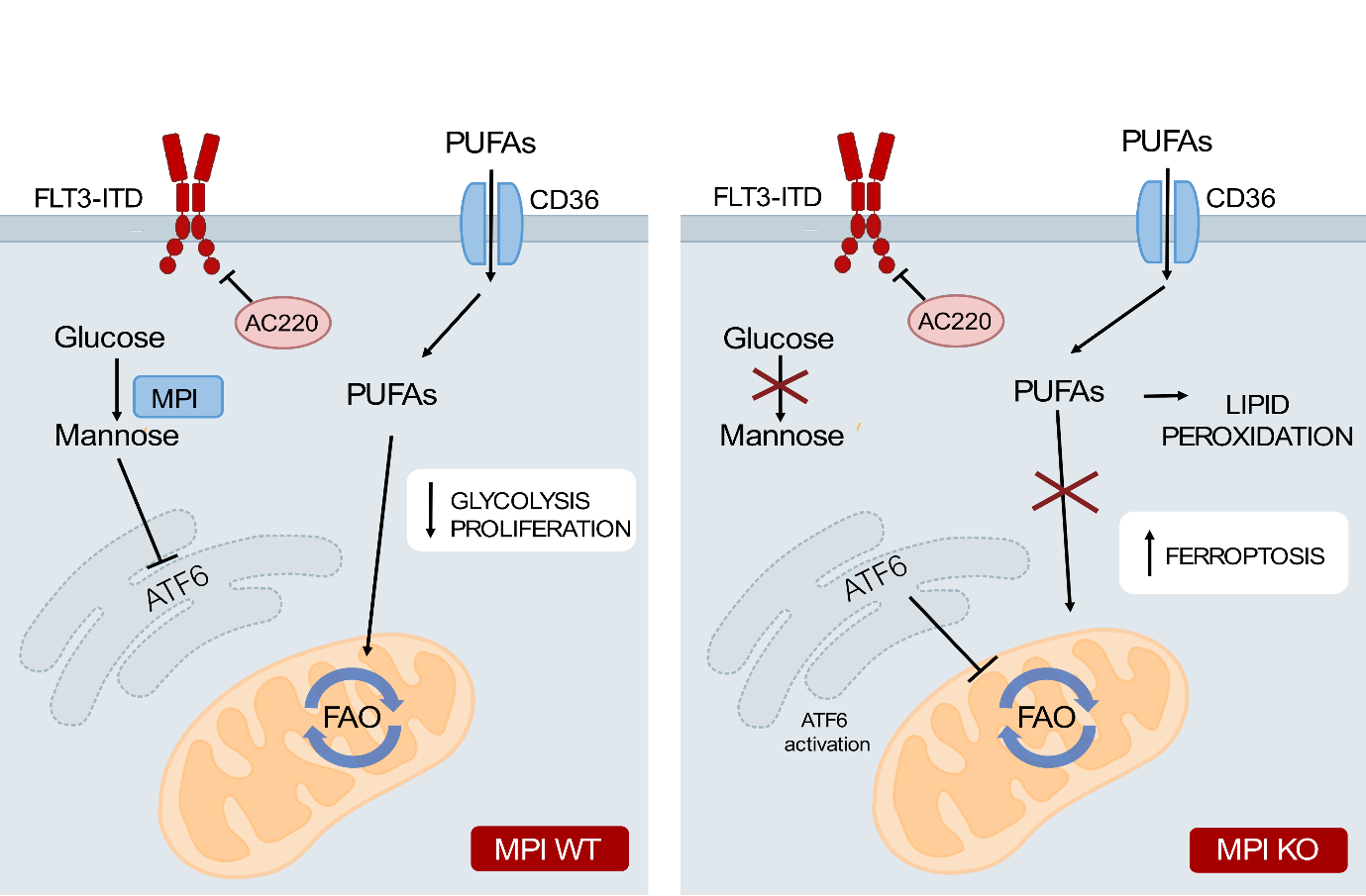
Loss of MPI leads to cell death in AML through inhibition of FAO leading to PUFA accumulation and ferroptosis.
Wild type AML cells treated with therapies are able to escape cell death by adapting their metabolism, in this case by switching from glycolysis to fatty acid oxidation. AML cells with inhibited or depleted MPI have activation of the unfolded proteins response which causes ATF6 activation, inhibiting fatty acid oxidation. This is paired with increased uptake of fatty acids, particularly polyunsaturated fatty acids, by MPI depleted cells. These PUFAs undergo lipid peroxidation which leads to ferroptotic cell death in these cells (Figure for ongoing project A and B).
Mannose metabolism inhibition sensitizes acute myeloid leukemia cells to cytarabine and FLT3 inhibitor therapy by modulating fatty acid metabolism to drive ferroptotic cell death.
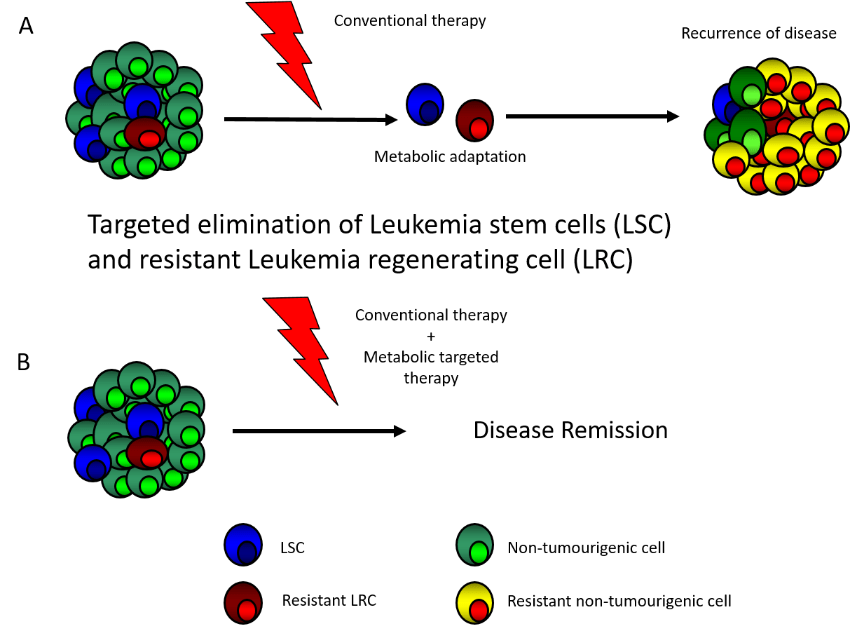
A) Standard AML therapy reduce disease bulk but do not eradicate resistant LRC and LSC which often utilise metabolic adaptations to survive and in turn lead to relapse. B) We aim to identify such adaptations and by targeting them promote durable remission and cure.
Collaborators
- Prof Cristina Lo Celso, Imperial College and Francis Crick Institute, London
- Dr Richard Burt, Francis Crick Institute, London
- Dr Mariia Yuneva, Francis Crick Institute, London
- Prof Brian Huntly, Wellcome-MRC Cambridge Stem Cell Institute, Cambridge
- Dr George Vassiliou, Wellcome Trust Sanger Institute/ Wellcome-MRC Cambridge Stem Cell Institute, Cambridge
- Dr Vignir Helgason, Institute of Cancer Sciences, University of Glasgow
- Prof John Al Copland, Mayo Clinic, US
Other Activities
- Member of the ASH Global Research Award Grant Subcommittee
- Member of UK, National Cancer Research Institute (NCRI) CML and AML clinical and scientific study group
- Member of the American Society for Hematology
- Member of the European Haematology association
- Trainee member of the UK, NCRI Haemato-Oncology Clinical Study Group - 2015-2016
Major Funding
- 2024 - 2025- Queen Mary Innovation Impact Fund, £19,840
- 2023 - 2026- Barts Charity project grant, £320,237
- 2023 - NIHR Academic Clinical Fellowship, "Identification of novel therapies in acute myeloid leukaemia (AML)", approx. £200,000
- 2022 - Cancer Research UK City of London Development Fund 2022, “Identification of the metabolic determinants of the anti-leukemic effects of stearoyl-coA desaturase (SCD) inhibition”, £ 25,000
- 2022 - Lady Tata Memorial Trust International Award (awarded to Dr Dembitz Postdoctoral Research Assistant in the lab), “Mechanisms of anti-leukemic effects of stearoyl-coA desaturase (SCD) inhibition”, £33,000
- 2021-2025 - CRUK City of London PhD Scholarship, "Exploring adaptive resistance to targeted therapy in AML," £146,622
- 2019-2024- CRUK Advanced Clinician Scientist Fellowship, "Identification and characterisation of metabolic pathways as determinants of drug resistance in acute myeloid leukaemia," £1,427,558
- 2018-2020- American Society of Hematology Global Research Award, "Preliminary investigation on the role of metabolic pathways as determinants of drug responses in acute myeloid leukemia," $140,000
- 2016-2019- Wellcome Trust Postdoctoral Research Training Fellowship for Clinicians, "The role of aberrant metabolism in the pathogenesis and therapy of Acute Myeloid Leukaemia (AML)," £393,467
- 2014-2015- Lady Tata Memorial Trust International Awards, "Preliminary investigations on the role of aberrant metabolism in the pathogenesis and therapy of Acute Myeloid Leukaemia (AML)," £30,000
- 2014-2016- The Academy of Medical Sciences, Starter Grant for Clinical Lecturers, "Investigation of the role of the CBP and p300 lysine acetyltransferases in the maintenance of acute myeloid leukaemia," £30,000
- 2010-2013- Medical Research Council, UK, Clinical Research Training Fellowship, "The relevance of autocrine growth factors activation to the survival and proliferation of primitive chronic myeloid leukaemia cells," £203,583
Recent Publications
Immunodeficient NBSGW mouse strain allows chemotherapy modeling in AML patient‐derived xenografts Dembitz V, Durko J, Campos J et al. HemaSphere (2024) 8(10)
Molecular MRD is strongly prognostic in patients with NPM1-mutated AML receiving venetoclax-based nonintensive therapy Othman J, Tiong IS, O'Nions J et al. Blood (2024) 143(10) 336-341
Real-World Effectiveness of Asciminib in Patients with Chronic Myeloid Leukemia (CML) Harboring the T315I Mutation: A Global Chart Review Study of Patients Treated in the Asciminib Managed Access Program (MAP) Milojkovic D, Blijlevens N, Kwong Y-L et al. Blood (2023) 142(10) 4541
S124: PHOSPHOPROTEOMICS ACCURATELY PREDICTS RESPONSES TO MIDOSTAURIN PLUS CHEMOTHERAPY IN TWO INDEPENDENT COHORTS OF FLT3 MUTANT-POSITIVE ACUTE MYELOID LEUKAEMIA Dokal A, Borek WE, Nobre L et al. HemaSphere 7(10) e3920765
O20 TARGETING THE DEFECTIVE COA PATHWAY TO IMPROVE ERYTHROPOIESIS IN SF3B1-MUTANT MDS-RS PATIENTS Philippe C, Mian S, Maniati E et al. Leukemia Research (2023) 128(10) 107133
Mannose metabolism inhibition sensitizes acute myeloid leukaemia cells to therapy by driving ferroptotic cell death Woodley K, Dillingh LS, Giotopoulos G et al. Nature Communications 14(10) 2132
The outcome of post-transplant asciminib in patients with chronic myeloid leukaemia Fernando F, Innes AJ, Claudiani S et al. Bone Marrow Transplantation (2023) 58(10) 826-828
C/EBPα Confers Dependence to Fatty Acid Anabolic Pathways and Vulnerability to Lipid Oxidative Stress-Induced Ferroptosis in FLT3-Mutant Leukemia. Sabatier M, Birsen R, Lauture L et al. Cancer Discovery (2023) 13(10) 1720-1747
Vitamin B5 and succinyl-CoA improve ineffective erythropoiesis in SF3B1-mutated myelodysplasia Mian SA, Philippe C, Maniati E et al. Science Translational Medicine (2023) 15(10) eabn5135-eabn5135
Inhibition of Stearoyl-CoA Desaturase Has Anti-Leukemic Properties in Acute Myeloid Leukemia Dembitz V, Lawson H, Philippe C et al. Blood (2022) 140(10) 3058-3060
For additional publications, please click hereTeam
Postdoctoral Researchers- Dr Sirisha Natani
- Dr Mike Williams
- Dr Samantha Mason
- Sophie James

Biography
I am a clinical academic haematologist with an interest in the biology and therapy of myeloid leukaemias. I studied Medicine in Naples, Italy, where I graduated in 2002. I then moved to the UK for my postgraduate medical training including my Haematology specialty training. I obtained my PhD, funded via an MRC Clinical Research Training Fellowship, in Prof Holyoake lab in Glasgow in 2013, studying resistance mechanism of Chronic Myeloid Leukaemia (CML) stem cells to BCR-ABL tyrosine kinase inhibitors. I was awarded the Royal College of Pathologist, UK Haematology Specialty Research Medal Award for this work. I then moved to Cambridge to complete my specialty training and worked as a Wellcome Trust funded Clinical Post-doctoral Fellow within Prof Brian Huntly lab where I have developed an independent research line focusing on the role of altered metabolism in the pathogenesis and therapy of myeloid leukaemias. I am clinically active at Consultant level with a leading role on design and set-up of clinical trials through my membership of the NCRI AML and CML Working Group. I am already a principal investigator/sub-investigator on several clinical trials in myeloid malignancies. I moved to BCI in 2019 to start my independent research programme which is primarily funded by a CRUK Advanced Clinician Scientist Fellowship. Qualifications- Certificate of Completion of Training (CCT) in Haematology, December 2014
- Fellowship Royal College of Pathologist in Haematology (FRCPath), UK, May 2014
- PhD in Experimental Haematology, University of Glasgow, UK, April 2013
- Membership Royal College of Physicians (MRCP), UK, July 2005
- Laurea in Medicina&Chirurgia (Degree in Medicine&Surgery) Grade: 110/110 summa cum laude awarded with distinction (top 5% of the class), University of Naples “Federico II”, Italy, July 2002